Citation
Guo, P.; Wang, B.; Bauchy, M.; Sant, G. Crystal Growth & Design 2016,16, no. 6: 3124-32.
Guo, P.; Wang, B.; Bauchy, M.; Sant, G. Crystal Growth & Design 2016,16, no. 6: 3124-32.
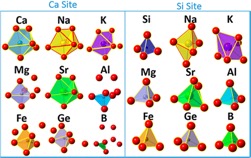
Density functional theory (DFT) simulations are carried out to systemically investigate doping in dicalcium silicate (Ca2SiO4: C2S), a major phase in calcium silicate cements. By evaluating the energetics of defect formation mechanisms for species involving Na+, K+, Mg2+, Sr2+, Al3+, Fe3+, B3+, and Ge4+, we find a strong site preference for all cationic substitutions. As a result, distinct defects form at low dopant concentrations (i.e., ≤ 0.52 atom %), in which, expectedly, larger dopants prefer (larger) Ca2+ sites, while smaller dopants favor (smaller) Si4+ sites, with charge balance being ensured by the formation of vacancies. Such site preferences arise due to local atomic distortions, which are induced when doping occurs at unfavorable substitution sites. Interestingly, we note that the formation enthalpy of each substitutional defect is proportional to the size mismatch between the dopant and the native cations. This indicates that the destabilization of the C2S structure has its origins in an “atomic size misfit” which develops while accommodating defects in the C2S lattice. The outcomes resulting from this work provide insights that are needed to select dopants to optimize cement performance by compositional design.